verfasst von Felix C. Goerigk, Institu für Anorganische Chemie der Universität Stuttgart
Vorwort
Über das Hinterlegen von Kristallstrukturen in der häufig benutzten ICSD gibt es ja gelegentlich Unklarheiten, insbesondere nach dem Zusammenschluss der ICSD mit der Cambridge Structural Database. Früher konnte ein fertiger Datensatz, bestehend aus .cif-Datei und .hkl-Datei einfach per Mail an das Fachinformationszentrum (FIZ) Karlsruhe geschickt werden. Seit dem Zusammenschluss wird dies nun Online über einen Assistenten gemacht. Dies ist zwar etwas umständlicher, hat aber den Vorteil, eventuelle Fehler in den Datensätzen noch aufzuzeigen, die sonst übersehen werden könnten. Da die ICSD mit eine der wichtigsten Strukturdatenbanken für anorganische Strukturen ist, soll es im Folgenden über das gängige Vorgehen des Strukturhinterlegens gehen.
Zur Hinterlegung einer neuen Kristallstruktur müssen folgende Voraussetzungen erfüllt werden:
- fertiger Kristallstruktur-Datensatz, bestehend aus .cif- , .hkl- und .fcf-Datei. Die .cif- und die .fcf-Datei wird in der Regel beim Verfeinern der Kristallstruktur durch die jeweilige Software ausgegeben. Als .hkl-Datei sollte die vom Diffraktometer bei der Integration erzeugte Datei verwendet werden, die anschließend noch eine Absorptionskorrektur, z.B. mit STOE X-SHAPE, Habitus oder dem Laue Analyser (LANA) durchlaufen hat. Die letzten genannten Programme sind kommerziell bei Stoe Darmstadt im Softwarepaket Stoe X-Area erhältlich bzw. bei Datenverarbeitung von Stoe-Diffraktometern üblich. Weitere Informationen kann Quelle [1] entnommen werden.
- Einen Account beim Cambridge Crystallographic Data Centre (CCDC). Dieser ist kostenlos einrichtbar.
- Die Software EnCIFer (nicht unbedingt erforderlich, erleichtert das Arbeiten aber deutlich). Diese ist beim CCDC ebenfalls kostenlos beziehbar. Alternativ kann auch der Online-Service genutzt werden.
Schritt-für-Schritt-Vorgehen
Überprüfen der .cif-Datei
Zunächst sollte die fertige .cif-Datei mittels des Programms EnCIFer geladen werden. Dieses Tool kennt erlaubte Befehle der .cif-Nomenklatur und gibt sofort Fehler aus, wenn beim manuellen Editieren unzulässige Einträge vorgenommen wurden. Natürlich kann einen beliebigen Texteditor verwenden, allerdings können dann Fehler leicht übersehen werden, da keine Rückmeldung zu durchgeführten Eingaben erfolgt. Der grundlegende Umgang mit .cif-Dateien wird im Folgenden vorausgesetzt.
In der folgenden Tabelle sind einige Einträge der Datei genannt, die besonders beachtet werden sollten, da hier in der Regel von Hand Änderungen nötig sein könnten. Außer den unten angegeben Werten, die in der Regel immer modifiziert werden müssen, sollten auch die bereits eingetragenen Gütewerte, Anzahl der Reflexe, Messbereich, berechnete Dichte etc. ebenfalls noch auf ihre Stimmigkeit kontrolliert werden. Je nach verwendeter Hard- und Software und Messgeometrie können manche Einträge auch nicht erforderlich sein.
| Bezeichnung des Eintrags | Bedeutung | Was ist zu tun? |
_chemical_formula_sum |
Summenformel der Verbindung mit alphabetischer Sortierung der Elemente | kontrollieren, ob korrekt |
_symmetry_cell_setting |
Kristallsystem |
eintragen, als ein Wort, z.B. monoclinic |
_symmetry_space_group_name_H-M |
Raumgruppentyp in Hermann-Mauguin-Symbolik |
eintragen, z.B. P21/c |
_cell_lengthund _cell_angle |
Zellparameter | kontrollieren. Es müssen immer alle drei Achsen und Winkel angegeben werden, unabhängig von der Symmetrie! |
_cell_measurement_theta_minund _cell_measurement_theta_max |
Minimal- und Maximalwert von θ (nicht 2θ) für die Zellbestimmung | Aus Messprotokoll des Diffraktometers ermitteln und eintragen |
_exptl_crystal_descriptionund _exptl_crystal_colour |
Habitus und Farbe des Kristalls |
eintragen, z.B. needle, bar, plate, und bei Farbe pale yellow, colourless, |
_exptl_crystal_size_maxund _exptl_crystal_size_midund _exptl_crystal_size_min |
maximale, mittlere und minimale Kantenlänge des Kristalls | Werte in Millimeter eintragen. Sinnvoll lassen sich diese aus licht- oder besser elektronen-mikroskopischen Aufnahmen des Kristalls ermitteln. |
_exptl_absorpt_correction_type |
Art der Absorptionskorrektur |
ergänzen, z.B. numerical oder multi-scan |
_exptl_absorpt_correction_T_minund _exptl_absorpt_correction_T_max |
minimaler und maximaler Transmissionskoeffizient | ergänzen, wird in der Regel durch die Software der Absorptions-korrektur berechnet. Erlaubte Werte gehen von 0.0 bis 1.0 |
_exptl_absorpt_process_details |
Verwendete Programme der Absorptionskorrektur | Programm, Version und Herausgeber eintragen, alle Werte in Hochkommas (‚Hochkomma‘) setzen. |
_diffrn_measurement_device_type |
Diffraktometermodell | Hersteller und Gerät eingeben, alles in Hochkommas |
_diffrn_measurement_method |
Geometrie und Detektor des Diffraktometers |
eingeben, z.B. ‚four-circle, ccd-detector’ alles in Hochkommas |
_computing_data_collectionund _computing_cell_refinementund _computing_data_reduction |
Software der Messung, Verfeinerung und Datenreduktion | Eingeben, alles in Hochkommas |
_computing_structure_solutionund _computing_structure_refinement |
Herausgeber und Version der Software | meist automatisch gesetzt, checken, in Hochkommas, z.B.
'SHELXL-97 (Sheldrick, 1997)' |
Anschließend wird in EnCIFer auf das Kontroll-Symbol (gelbes Warndreieck mit zwei Pfeilen) geklickt. Im Idealfall wird unten „Errors – none“, „Warnings – none“ und „remarks – none“ angezeigt. In der Abbildung 1 wurde hingegen absichtlich ein Fehler eingebaut. Die aus den Symmetrieoperationen richtige Raumgruppe wäre P21/c, EnCIFer hebt die falsch eingegebene Raumgruppe P2/c hervor (bei „Warnings“).

Hinterlegen mit Hilfe des Online-Assistenten
Nachdem die .cif-Datei fehlerfrei ist, kann die Struktur hinterlegt werden. Um sie in der ICSD zu hinterlegen, muss sie erst in der CCDC hinterlegt werden. Klingt komisch, ist aber aufgrund des Zusammenschlusses so. Hierzu legt man sich, falls noch nicht vorhanden, ein Profil an, klickt nach Einloggen in der CCDC auf „Profile“ und anschließend links im Menü auf „Deposit“. Der Assistent ist an sich relativ gut erklärt. Zunächst sollten die persönlichen Daten kontrolliert werden, anschließend werden unter „Select Files“ die .cif-, .hkl- und .fcf-Datei hochgeladen. Unten kann noch die Option „I wish to run the IUCr checkCIF/PLATON service on my data” ausgewählt werden, hierbei wird die Datenbank auf bereits vorhandene, identische Metriken sowie auf Fehler in der .cif-Datei untersucht. Sollte „View Report“ orange oder rot aufleuchten, so sind offenbar Fehler aufgetreten, die dann angezeigt und korrigiert werden sollten. Diese sind in verschiedene „Alert Levels“ kategorisiert, wobei Level A und B in der Regel ernste Probleme bedeuten (Tippfehler in der .cif-Datei?) und z.B. Level G nur allgemeine Anmerkungen sind. Die Temperatur, bei der gemessen wurde, wird oft als Level-C-Fehler angezeigt. Dieser Hinweis kann nach Kontrolle ignoriert werden.
Sind alle Probleme beseitigt, kann auf die nächste Seite „Add Publication“ gegangen werden. Im weiteren Verlauf muss dann noch die Literaturstelle angegeben werden, in der die Verbindung erstmals publiziert wurde. Da man aber im Normalfall erst hinterlegt und dann publiziert, werden hier zunächst vorübergehend nur die Namen und Adressdaten aller geplanter Autoren eingetragen. Das Journal und die genaue Seitenzahl werden dann später nach Publikation einfach ergänzt.
Auf den folgenden Seiten „Enhance Data“ und „Review“ werden die eingegeben Daten nochmals zusammenfassend dargestellt. Hier können auch noch katalogisierende Einträge angegeben werden, die ein schnelleres Suchen und Finden für andere Benutzer erleichtern. Diese sind rechts ankreuzbar und sollten so genau wie möglich beantwortet werden. Bei einer festkörperchemisch aus einer Salzschmelze hergestellten Verbindung mit Lumineszenzeigenschaften, sollten beispielsweise die Eigenschaften „from the melt“ und „Luminescent“ ausgewählt werden. Weitere, selbsterklärende Optionen z.B. zu der Stabilität sind ebenfalls auswählbar. Nachdem man zufrieden ist, kann der Datensatz mit „Submit“ eingereicht werden. Innerhalb weniger Minuten sollte eine automatische Mail mit der CSD-Nummer eingehen, ferner kann der Datensatz unter „Profile“ begutachtet werden.
Weitere Publikation und Verlinken mit der ICSD
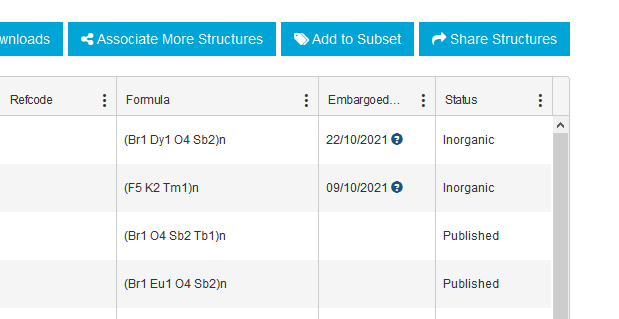
Unter „My structures“ in “Profile” findet sich eine tabellarische Auflistung aller hinterlegter Datensätze. Sind diese noch mit keiner Literatur verknüpft, so findet sich wie in Abbildung 2 dargestellt ein Embargo-Datum. Dies ist in der Regel ein Jahr nach dem Tag der Hinterlegung. Bis zu diesem Zeitpunkt sollte eine Literaturstelle angegeben werden, da die Verbindung ansonsten nicht mehr direkt mit der Veröffentlichung assoziiert werden kann. Zur Verknüpfung wird auf „Details“ in der Zeile der entsprechenden Verbindung geklickt, auf der nächsten Seite unten dann auf „Add Publication“ geklickt, wo die entsprechenden Angaben eingetragen werden. Nach Prüfung der Eingaben stellt sich der Status auf „Published“ um.
Man könnte meinen, nun fertig zu sein und alles ist in Ordnung. Leider ist noch ein letzter Schritt erforderlich, der nicht erklärt wird: Nun muss nämlich noch die Verlinkung zur ICSD vorgenommen werden. Leider funktioniert dies nicht automatisch. Um die fertige Struktur auch in die ICSD aufzunehmen, wird manuell von der Dienst-Adresse (bzw. der Adresse, die im CCDC-Profil eingetragen wurde) eine Mail an CrysDATA@fiz-karlsruhe.de geschrieben, die neben der Nennung des Accounts und der Verbindung (CSD-Nummer) auch eine .pdf-Datei des publizierten Papers (fertige Version inklusive Seitenzahlen, keine Druckfahne etc.) enthält. Dann wird in der Regel der Eingang der Referenz per Mail bestätigt und die Verbindung aus der CCDC in die ICSD beim nächsten Update eingefügt.
Warum man hier von Hand noch eine Mail mit dem Paper an das FIZ schicken muss, ist mir schleierhaft. Der Online-Assistent bei der CCDC funktioniert ausgesprochen gut, es scheint nur ein Kommunikationsproblem zwischen der CCDC und dem FIZ zu geben, da das FIZ gemäß eigener Aussage keine Informationen über die Publikationsdetails von der CCDC erhält und daher nicht automatisch sieht, wann eine Verbindung auch tatsächlich ordnungsgemäß veröffentlicht wurde. Dieser letzte Schritt sollte noch verbessert und automatisiert werden. Unternimmt man nichts weiter, bleibt die Verbindung in der CCDC zwar „Published“, wird aber nicht in die ICSD aufgenommen.
Ich hoffe, der Beitrag hilft dem einen oder anderen weiter, die eigenen erfolgreich Strukturen zu hinterlegen!
Mit Grüßen aus Stuttgart,
Felix C. Goerigk
Felix Goerigk promovierte im Arbeitskreis von Prof. Thomas Schleid am Institut für Anorganische Chemie der Universität Stuttgart. Seine Forschungsschwerpunkte umfassen die Strukturaufklärung neuer Verbindungen mittels Einkristall- und Pulverröntgenbeugung und angrenzender Methoden wie REM-EDS und -WDS.
[1] W. Herrendorf, H. Bärnighausen, HABITUS: Programm zur Optimierung der Kristallgestalt für die numerische Absorptionskorrektur als Version X-SHAPE (Version 1.06, Fa. Stoe, Darmstadt 1999). Karlsruhe, Gießen, 1996.