Written by Tatjana Barthel Macromolecular Crystallography group at BESSY II)
The outbreak of SARS-CoV-2 pushed all of us into a very complicated situation, especially with strict regulations and certain lockdown periods. Performing science became challenging in these last weeks, but at the same time it is of great importance in order to fight the virus. In the development of drugs against SARS-CoV-2 X-ray crystallography plays a key role.
One field of X-ray crystallography is protein crystallography. Proteins can form crystals just as small molecules, metals and salts. The array of ordered protein molecules inside a protein crystal leaves gaps that are filled by disordered solvent molecules, i.e. mostly water. These so called solvent channels enable small molecules to enter the protein crystals and reach the individual protein molecules. Thus, protein crystals can be used for drug development, by probing them with small molecules in order to find out if and how the molecules bind to the protein. The discovered interactions between the protein and such small molecules can be used as starting points to design efficient drugs. X‑ray crystallography can be applied throughout the whole process of finding start molecules and iteratively optimizing them into drug candidates. Many labs already started such a process in connection to SARS-CoV-2, to find molecules that bind to certain proteins of the virus and thus can be optimized to hamper virus reproduction or infectivity. A prominent protein target is the main protease MPro, an enzyme of SARS-CoV-2 that is important for the replication of the virus. Therefore, by impeding the function of MPro, the virus will no longer be able to multiply. In order to find appropriate starting points for the development of drugs via X-ray crystallography, different approaches are being applied by different labs at synchrotron sites, for instance at DIAMOND light source [1], PETRA III [2] and BESSY II.
I am Tatjana Barthel, a PhD student in the Macromolecular Crystallography group at BESSY II and I take part in one of our MPro targeting projects. The project is a collaboration of the group of Rolf Hilgenfeld at Universität zu Lübeck, who discovered the structure of the MPro protein [3] and is coordinated on our side by our postdoc Jan Wollenhaupt. We chose to screen fragments, i.e. small molecules usually below 300 Dalton (SI unit for atomic mass, 1 Dalton ~1.66 x 10-27 kg), using our newly developed fragment library the F2X-Entry Screen (comprised of 96 fragments). (Figure 1) Screening fragments has a great advantage as they are very promising starting points in order to design potent drugs. I also took part in the validation of this novel library in context of my Master Thesis, and we recently published those results [4]. Therefore, I knew already how to handle hundreds of protein crystals and applied these skills in the MPro project. The first experiments with MPro are now completed and we could identify fragments binding to a central part of MPro.
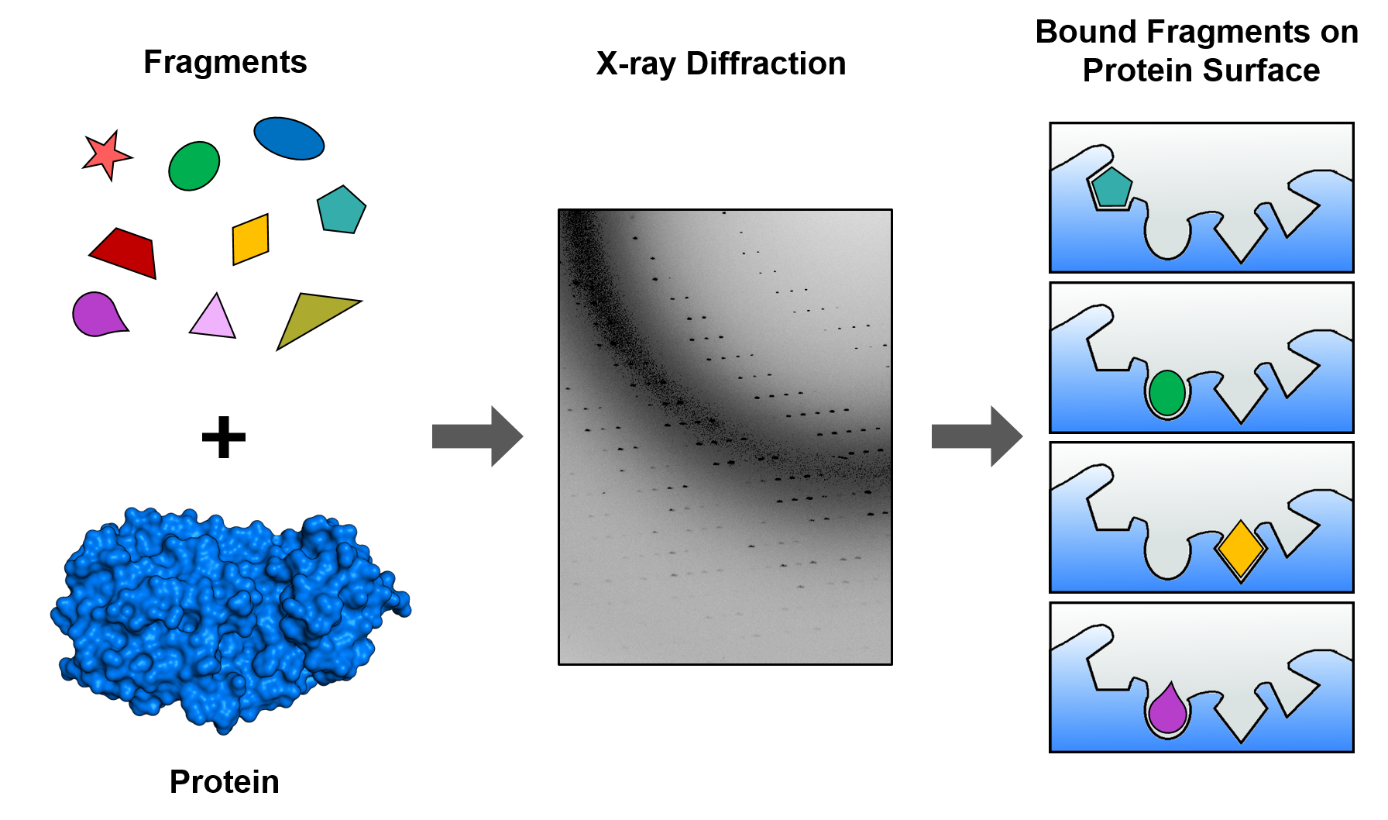
That means we have some interesting starting points, which are now used in a computational workflow to find larger molecules that present additional molecule-protein interactions and therefore are expected to bind stronger and thus have a bigger effect on the protein. These computational results will thereafter be validated again by X-ray crystallography. The goal is to do several optimization i.e. “molecule-growing” steps in order to arrive at molecules that are suitable for further screening in biological experiments to test their effect on the virus and proceed with optimized candidates further along the long path to create a SARS-CoV-2 drug.
Luckily, many scientists unite in the efforts of drug discovery. Another project coordinated by Christian Feiler of our group and the Berlin-based biotech company Molox is a large consortium of regional and international academic groups and large industrial partners like BASF. Their goal is to target many more SARS-CoV-2 proteins with X-ray crystallography assisted screening to identify further starting points for the development of a potential active substance against the coronavirus.
There are still many steps in front of all of us working on drugs against SARS-CoV-2, but hopefully we can all progress as fast as possible.
References:
[1] A. Douangamath et al., “Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease,” bioRxiv, p. 2020.05.27.118117, May 2020, doi: 10.1101/2020.05.27.118117.
[2] S. Günther et al., “Catalytic cleavage of HEAT and subsequent covalent binding of the tetralone moiety by the SARS-CoV-2 main protease,” biorxiv, p. 2020.05.02.043554, May 2020, doi: 10.1101/2020.05.02.043554.
[3] L. Zhang et al., “Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors,” Science (80-. )., vol. 368, no. 6489, p. 409, Apr. 2020, doi: 10.1126/science.abb3405.
[4] J. Wollenhaupt and A. Metz et al., “F2X-Universal and F2X-Entry: Structurally Diverse Compound Libraries for Crystallographic Fragment Screening,” Structure, May 2020, doi: 10.1016/j.str.2020.04.019.